Loading included data
You can load the demo data in microbiomedataset package to see the microbiome_dataset class.
library(microbiomedataset)
library(tidyverse)
data("global_patterns")
global_patterns
#> --------------------
#> microbiomedataset version: 0.99.1
#> --------------------
#> 1.expression_data:[ 19216 x 26 data.frame]
#> 2.sample_info:[ 26 x 8 data.frame]
#> 3.variable_info:[ 19216 x 8 data.frame]
#> 4.sample_info_note:[ 8 x 2 data.frame]
#> 5.variable_info_note:[ 8 x 2 data.frame]
#> --------------------
#> Processing information (extract_process_info())
#> create_microbiome_dataset ----------
#> Package Function.used Time
#> 1 microbiomedataset create_microbiome_dataset() 2022-07-10 10:56:13
So you can see that we have 1,9216 variables and 26 samples in the dataset.
Create microbiome_dataset class object
You can also create the microbiome_dataset class using the create_microbiome_dataset function.
We need to prepare at least three data for it.
-
expression_data: rows are variables and columns are samples. -
sample_info: Information for all the samples inexpression_data. The first column should besample_idwhich should be identical with the column names ofexpression_data. -
variable_info: Information for all the variables inexpression_data. The first column should bevariable_idwhich should be identical with the row names ofexpression_data.
expression_data <-
as.data.frame(matrix(
sample(1:100, 100, replace = TRUE),
nrow = 10,
ncol = 10
))
rownames(expression_data) <- paste0("OTU", 1:nrow(expression_data))
colnames(expression_data) <-
paste0("Sample", 1:ncol(expression_data))
expression_data
#> Sample1 Sample2 Sample3 Sample4 Sample5 Sample6 Sample7 Sample8 Sample9
#> OTU1 89 91 32 7 70 79 79 48 52
#> OTU2 40 59 83 58 71 78 17 90 27
#> OTU3 15 71 74 37 58 64 75 66 7
#> OTU4 100 13 49 10 50 88 97 52 70
#> OTU5 81 47 14 17 84 66 35 16 68
#> OTU6 11 95 76 82 59 79 67 88 56
#> OTU7 28 3 88 4 33 78 15 13 64
#> OTU8 67 54 58 69 28 66 99 20 28
#> OTU9 13 91 87 30 94 40 23 18 88
#> OTU10 65 87 71 21 6 46 29 59 50
#> Sample10
#> OTU1 32
#> OTU2 3
#> OTU3 17
#> OTU4 59
#> OTU5 82
#> OTU6 95
#> OTU7 87
#> OTU8 35
#> OTU9 13
#> OTU10 100
variable_info <-
as.data.frame(matrix(
sample(letters, 70, replace = TRUE),
nrow = nrow(expression_data),
ncol = 7
))
rownames(variable_info) <- rownames(expression_data)
colnames(variable_info) <-
c("Domain",
"Phylum",
"Class",
"Order",
"Family",
"Genus",
"Species")
variable_info$variable_id <-
rownames(expression_data)
variable_info <-
variable_info %>%
dplyr::select(variable_id, dplyr::everything())
sample_info <-
data.frame(sample_id = colnames(expression_data),
class = "Subject")
object <-
create_microbiome_dataset(
expression_data = expression_data,
sample_info = sample_info,
variable_info = variable_info
)
object
#> --------------------
#> microbiomedataset version: 0.99.1
#> --------------------
#> 1.expression_data:[ 10 x 10 data.frame]
#> 2.sample_info:[ 10 x 2 data.frame]
#> 3.variable_info:[ 10 x 8 data.frame]
#> 4.sample_info_note:[ 2 x 2 data.frame]
#> 5.variable_info_note:[ 8 x 2 data.frame]
#> --------------------
#> Processing information (extract_process_info())
#> create_microbiome_dataset ----------
#> Package Function.used Time
#> 1 microbiomedataset create_microbiome_dataset() 2023-09-15 21:07:52
Convert phyloseq class to microbiome_dataset class object
We can also transfer or convert other common class object to microbiome_dataset class.
Please install phyloseq package at first.
if(!require(BiocManager)){
install.packages("BiocManager")
}
if(!require(phyloseq)){
BiocManager::install("phyloseq")
}
library(phyloseq)
data(GlobalPatterns)
GlobalPatterns
#> phyloseq-class experiment-level object
#> otu_table() OTU Table: [ 19216 taxa and 26 samples ]
#> sample_data() Sample Data: [ 26 samples by 7 sample variables ]
#> tax_table() Taxonomy Table: [ 19216 taxa by 7 taxonomic ranks ]
#> phy_tree() Phylogenetic Tree: [ 19216 tips and 19215 internal nodes ]
The first function is convert2microbiome_dataset:
object1 <-
convert2microbiome_dataset(object = GlobalPatterns)
object1
#> --------------------
#> microbiomedataset version: 0.99.1
#> --------------------
#> 1.expression_data:[ 19216 x 26 data.frame]
#> 2.sample_info:[ 26 x 8 data.frame]
#> 3.variable_info:[ 19216 x 8 data.frame]
#> 4.sample_info_note:[ 8 x 2 data.frame]
#> 5.variable_info_note:[ 8 x 2 data.frame]
#> --------------------
#> Processing information (extract_process_info())
#> create_microbiome_dataset ----------
#> Package Function.used Time
#> 1 microbiomedataset create_microbiome_dataset() 2023-09-15 21:08:03
The second function is as.microbiome_dataset:
object2 <-
as.microbiome_dataset(object = GlobalPatterns)
object2
#> --------------------
#> microbiomedataset version: 0.99.1
#> --------------------
#> 1.expression_data:[ 19216 x 26 data.frame]
#> 2.sample_info:[ 26 x 8 data.frame]
#> 3.variable_info:[ 19216 x 8 data.frame]
#> 4.sample_info_note:[ 8 x 2 data.frame]
#> 5.variable_info_note:[ 8 x 2 data.frame]
#> --------------------
#> Processing information (extract_process_info())
#> create_microbiome_dataset ----------
#> Package Function.used Time
#> 1 microbiomedataset create_microbiome_dataset() 2023-09-15 21:08:12
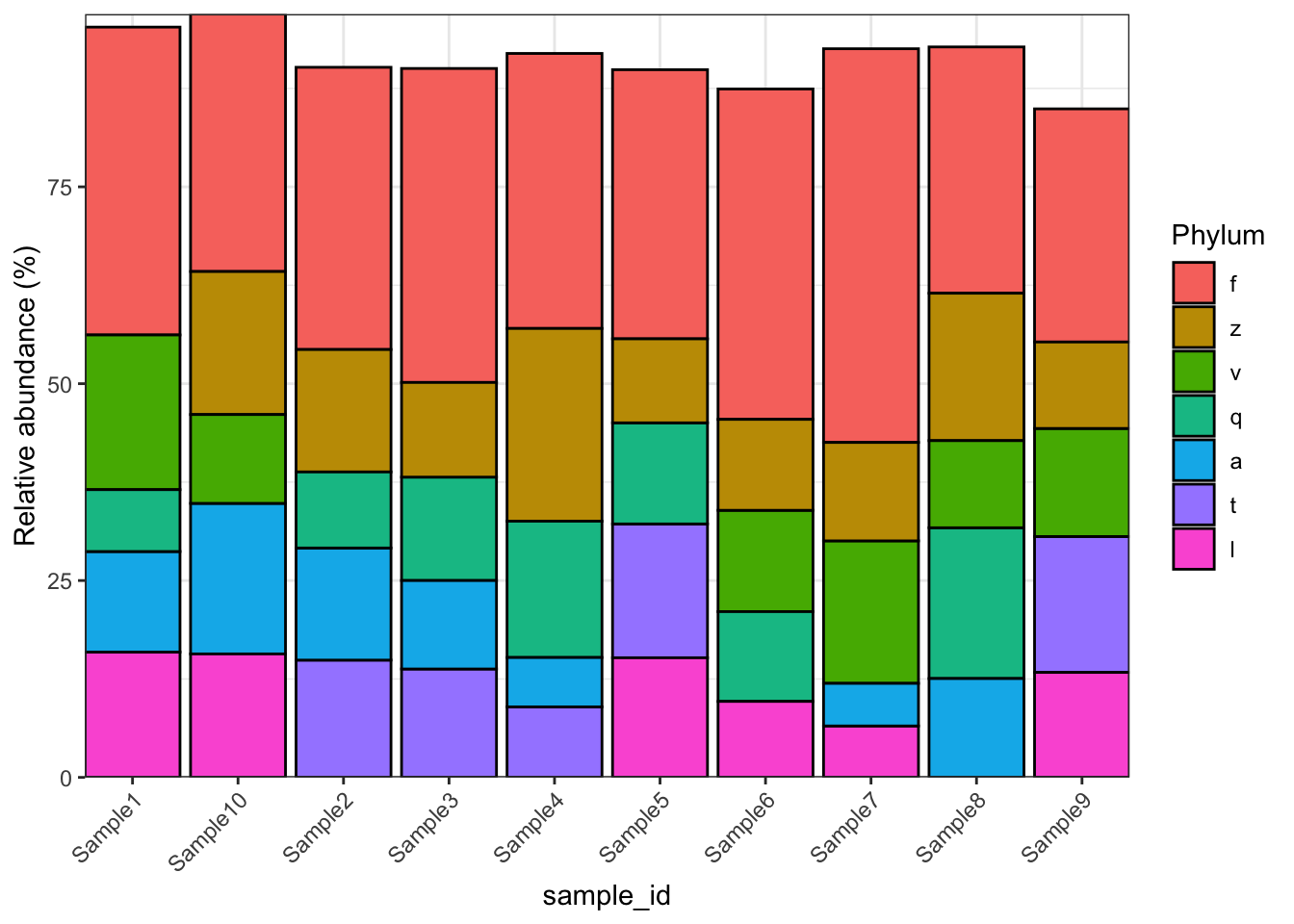
microbiomedataset::plot_barplot(object = object,
top_n = 5,
fill = "Phylum")
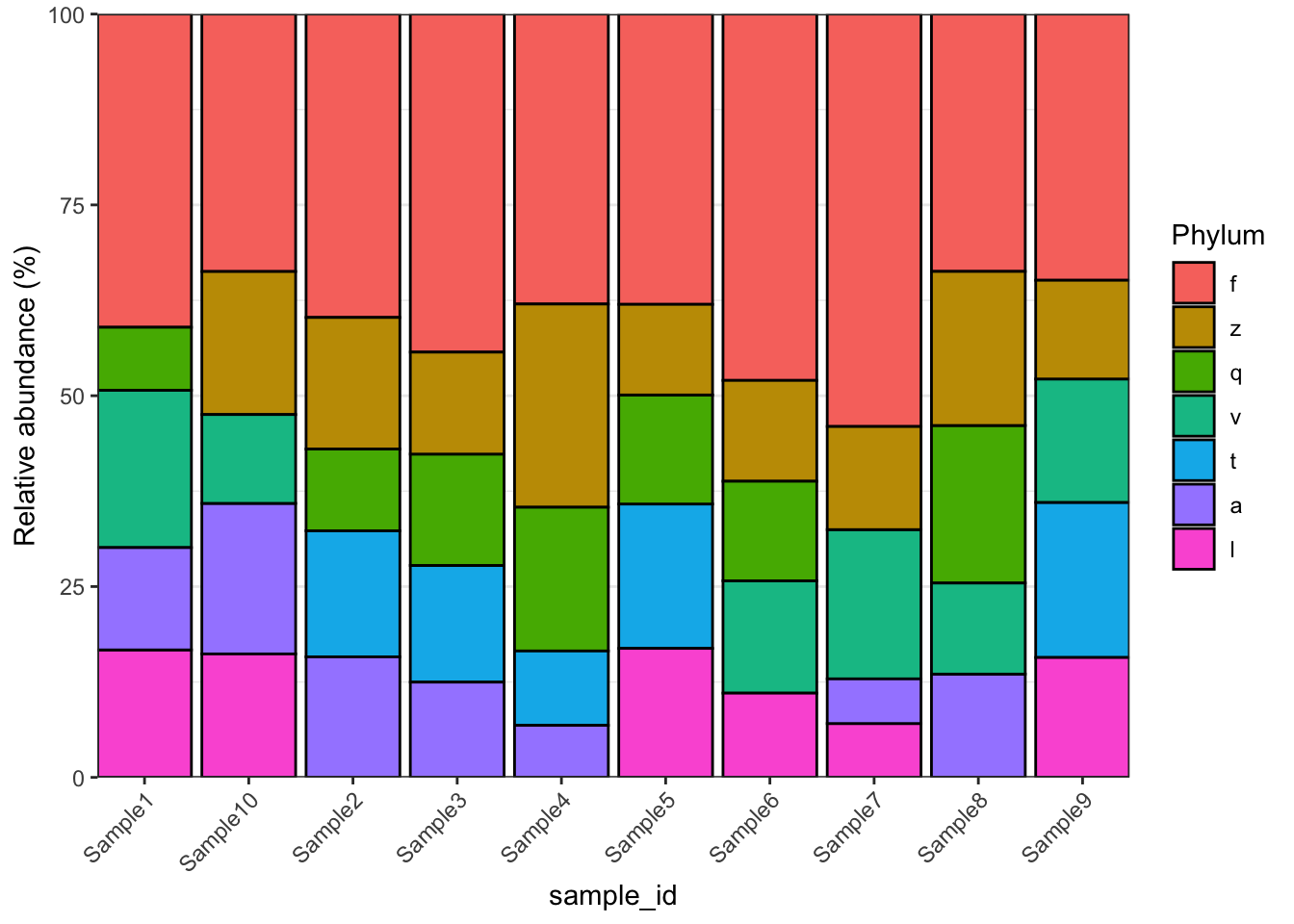
microbiomedataset::plot_barplot(object = object,
top_n = 5,
fill = "Phylum",
relative = TRUE,
re_calculate_relative = TRUE)
Session information
sessionInfo()
#> R version 4.3.0 (2023-04-21)
#> Platform: x86_64-apple-darwin20 (64-bit)
#> Running under: macOS Ventura 13.5.2
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: America/Los_Angeles
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] phyloseq_1.44.0 BiocManager_1.30.21
#> [3] lubridate_1.9.2 forcats_1.0.0
#> [5] stringr_1.5.0 dplyr_1.1.2
#> [7] purrr_1.0.1 readr_2.1.4
#> [9] tidyr_1.3.0 tibble_3.2.1
#> [11] ggplot2_3.4.2 tidyverse_2.0.0
#> [13] microbiomedataset_0.99.10
#>
#> loaded via a namespace (and not attached):
#> [1] RColorBrewer_1.1-3 rstudioapi_0.14
#> [3] jsonlite_1.8.5 shape_1.4.6
#> [5] magrittr_2.0.3 farver_2.1.1
#> [7] MALDIquant_1.22.1 rmarkdown_2.22
#> [9] GlobalOptions_0.1.2 zlibbioc_1.46.0
#> [11] vctrs_0.6.2 multtest_2.56.0
#> [13] RCurl_1.98-1.12 blogdown_1.18.1
#> [15] htmltools_0.5.5 S4Arrays_1.0.4
#> [17] Rhdf5lib_1.22.0 rhdf5_2.44.0
#> [19] gridGraphics_0.5-1 mzID_1.38.0
#> [21] sass_0.4.6 bslib_0.5.0
#> [23] htmlwidgets_1.6.2 plyr_1.8.8
#> [25] zoo_1.8-12 plotly_4.10.2
#> [27] impute_1.74.1 cachem_1.0.8
#> [29] igraph_1.4.3 lifecycle_1.0.3
#> [31] iterators_1.0.14 pkgconfig_2.0.3
#> [33] Matrix_1.5-4 R6_2.5.1
#> [35] fastmap_1.1.1 GenomeInfoDbData_1.2.10
#> [37] MatrixGenerics_1.12.2 clue_0.3-64
#> [39] digest_0.6.31 pcaMethods_1.92.0
#> [41] colorspace_2.1-0 masstools_1.0.10
#> [43] S4Vectors_0.38.1 rprojroot_2.0.3
#> [45] GenomicRanges_1.52.0 vegan_2.6-4
#> [47] labeling_0.4.2 timechange_0.2.0
#> [49] fansi_1.0.4 httr_1.4.6
#> [51] mgcv_1.8-42 polyclip_1.10-4
#> [53] compiler_4.3.0 here_1.0.1
#> [55] remotes_2.4.2 withr_2.5.0
#> [57] doParallel_1.0.17 BiocParallel_1.34.2
#> [59] viridis_0.6.3 highr_0.10
#> [61] ggforce_0.4.1 MASS_7.3-58.4
#> [63] DelayedArray_0.26.3 biomformat_1.28.0
#> [65] rjson_0.2.21 permute_0.9-7
#> [67] ggsci_3.0.0 mzR_2.34.0
#> [69] tools_4.3.0 ape_5.7-1
#> [71] zip_2.3.0 glue_1.6.2
#> [73] rhdf5filters_1.12.1 nlme_3.1-162
#> [75] grid_4.3.0 cluster_2.1.4
#> [77] reshape2_1.4.4 ade4_1.7-22
#> [79] generics_0.1.3 gtable_0.3.3
#> [81] tzdb_0.4.0 preprocessCore_1.62.1
#> [83] data.table_1.14.8 hms_1.1.3
#> [85] tidygraph_1.2.3 utf8_1.2.3
#> [87] XVector_0.40.0 BiocGenerics_0.46.0
#> [89] ggrepel_0.9.3 foreach_1.5.2
#> [91] pillar_1.9.0 yulab.utils_0.0.6
#> [93] limma_3.56.2 splines_4.3.0
#> [95] circlize_0.4.15 tweenr_2.0.2
#> [97] lattice_0.21-8 survival_3.5-5
#> [99] tidyselect_1.2.0 ComplexHeatmap_2.16.0
#> [101] Biostrings_2.68.1 pbapply_1.7-0
#> [103] knitr_1.43 gridExtra_2.3
#> [105] bookdown_0.34 IRanges_2.34.0
#> [107] ProtGenerics_1.32.0 SummarizedExperiment_1.30.2
#> [109] Rdisop_1.60.0 stats4_4.3.0
#> [111] xfun_0.39 graphlayouts_1.0.0
#> [113] Biobase_2.60.0 MSnbase_2.26.0
#> [115] matrixStats_1.0.0 stringi_1.7.12
#> [117] lazyeval_0.2.2 yaml_2.3.7
#> [119] evaluate_0.21 codetools_0.2-19
#> [121] ggraph_2.1.0 MsCoreUtils_1.12.0
#> [123] ggplotify_0.1.0 cli_3.6.1
#> [125] affyio_1.70.0 munsell_0.5.0
#> [127] jquerylib_0.1.4 Rcpp_1.0.10
#> [129] GenomeInfoDb_1.36.0 png_0.1-8
#> [131] XML_3.99-0.14 parallel_4.3.0
#> [133] bitops_1.0-7 tidytree_0.4.2
#> [135] viridisLite_0.4.2 scales_1.2.1
#> [137] affy_1.78.0 openxlsx_4.2.5.2
#> [139] ncdf4_1.21 crayon_1.5.2
#> [141] GetoptLong_1.0.5 rlang_1.1.1
#> [143] massdataset_1.0.25 vsn_3.68.0